Os modelos poligénicos não se importam com o número de loci envolvidos numa determinada variação contínua, e no entanto esses loci existem e podem até fazer-se estimativas do seu número mínimo (cf. experiência de East no início deste capítulo). Os loci envolvidos na variação contínua são chamados em inglês "Quantitative Trait loci", abreviadamente QTL. Em muitos casos tem-se verificado que o seu número pode chegar às centenas, muitos deles pertencendo forçosamente a grupos de ligação comuns; pode acontecer que QTL do mesmo grupo de ligação se encontrem demasiado próximos para ser possível separar os seus efeitos fenotípicos (cf. "pseudo-alelos"); daí que se possa preferir referirem-se os QTL não individualmente mas em termos de factores efectivos, regiões cromossómicas contendo um ou mais QTL e segregando independentemente doutros factores efectivos. A capacidade de separar as localizações de QTL ligados entre si depende da densidade de marcadores cromossómicos disponíveis, e do poder estatístico que se consegue na amostragem das populações segregantes.
De qualquer modo, o mero cálculo do número de factores efectivos é obviamente insatisfatório; na medida do possível, pretende-se o conhecimento da sua localização e da sua expressão. Graças à possibilidade de isolar segmentos de DNA de qualquer organismo, tem sido possível utilizar muitos novos marcadores cromossómicos cujos fenótipos são demonstráveis por electroforese, o que, apoiado num desenho experimental adequado, tem permitido um tratamento mendeliano de cada QTL. O mapeamento de QTL com esses marcadores cromossómicos já demonstrou, em vários casos, que a variação genotípica em caracteres de variação contínua se distribui por quase todos os grupos de ligação, confirmando as observações clássicas, onde todos os tipos de aneuplóides apresentavam aberrações fenotípicas (por exemplo em Datura ou Triticum).
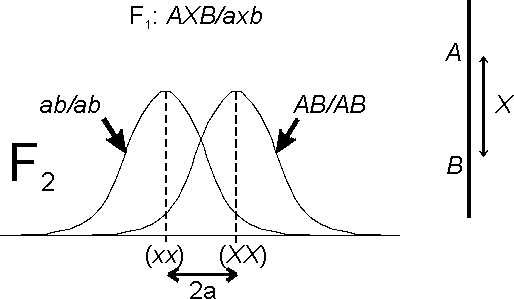
O mapeamento de QTL baseia-se na co-segregação de marcadores cromossómicos conjuntamente com variações fenotípicas significativas. Assim, diz-se que uma região cromossómica contém um QTL porque os marcadores dessa região estão associados a diferenças fenotípicas significativas que não se observam com os marcadores doutras regiões. Por exemplo (figura 17), se se cruzarem duas linhas puras com diferentes valores fenotípicos médios, produzindo-se uma F1 heterozigótica para diversos marcadores, diz-se que existe um QTL na região delimitada pelos loci marcadores, A e B, se na F2 se observar uma diferença significativa, no valor fenotípico médio, consoante os indivíduos são homozigóticos para o haplótipo AB ou para o ab (neste exemplo, a F1 é AB/ab). A diferença fenotípica entre esses homozigóticos permite ainda determinar aproximadamente o desvio aditivo correspondente a esse QTL.
Figura 17 — Exemplo dum QTL X/x, situado na região cromossómica delimitada pelos marcadores A e B, heterozigóticos na F1, em proximidade tal que a maior parte dos indivíduos XX e xx na F2 são identificados através dos homozigóticos nesses marcadores, assim podendo separar-se as respectivas distribuições e inclusivamente estimar o desvio aditivo a para este QTL.
O passo seguinte é tentar identificar cada locus cujos alelos contribuem para a variação contínua. Uma vez determinada com razoável precisão a localização cromossómica dum QTL, os loci que já se conheça nessa mesma região podem ser estudados um a um; mas é uma tarefa que pelas suas dimensões se pode revelar proibitiva, pelo número muito elevado de genes que existem na região e pelo esforço necessário para analisar cada um. Uma estratégia alternativa baseia-se em seleccionar a priori um locus "candidato", que pelo conhecimento sobre a função que codifica possa considerar-se um plausível componente da variação contínua do fenótipo em causa. Se esse locus se encontrar na vizinhança dum QTL, isso pode ser um argumento adicional a favor da hipótese que se coloca, e evidentemente um tal locus tem de ter pelo menos dois alelos nas populações onde se estuda essa variação, mas são apenas pontos de partida num trabalho muito complexo de demonstração da causalidade ontogénica e fisiológica para a variação, e que pode revelar-se frustrada se o locus candidato não for o QTL que se busca.
Contudo, é possível que muitos dos QTL tenham funções insuspeitadas ou mesmo desconhecidas, ou que uma função já conhecida tenha, duma maneira imprevista, um efeito pleiotrópico sobre a variação que se estuda. Por isso a estratégia dos loci candidatos só resulta em alguns casos. De facto, com os genomas que vão ficando sequenciados têm-se revelado muitas sequências nucleotídicas que aparentam ser genes funcionais, mas para as quais não se conhecem quaisquer funções ou proteínas conhecidas. Evidentemente que os QTL se contam entre os genes que vão sendo sequenciados, e o futuro permitirá por certo identificá-los pela função e pela posição dentro dos respectivos genomas; se alguns desses genes "desconhecidos" correspondem ou não a QTL, o futuro o dirá.